
DOI:https://doi.org/10.1007/s40145-022-0619-x
全文速览:
绿色太阳能驱动的光催化CO2还原是实现碳中和的一种很有前途的方法。在这项工作中,开发了一种新的缺陷工程方法,通过强大的水热过程,将SnO2重取代Nb掺杂形成SnxNb1−xO2固溶体。详细分析表明,Sn4+被高价位的Nb5+重取代后,由于其表面存在丰富的酸中心和多余的电子,形成了更合适的能带结构、更好的光生载流子分离和转移,并且具有更强的CO2吸附能力。因此,在不需要牺牲剂的情况下,SnxNb1−xO2固溶体样品比原始SnO2样品具有更好的光催化CO2还原性能。其光催化CO2还原效率达到~292.47µmol/(g·h),是原始SnO2样品的19倍。此外,其主要光催化CO2还原产物是C2H5OH的多碳(C2+)化合物,而原始SnO2样品的光催化CO2还原产物是CH3OH的一碳(C1)化合物。本研究表明,在金属氧化物中大量掺杂高价阳离子形成固溶体可以增强光催化CO2还原,并调节其还原过程,产生更多的C2+产物。这种材料设计策略可以很容易地应用于各种材料体系,用于探索太阳能驱动的二氧化碳减排的高性能光催化剂。
背景介绍:
本篇文章的研究背景是光催化二氧化碳还原反应。随着全球能源需求的增加和气候变化的日益严重,利用太阳能驱动的二氧化碳还原反应将二氧化碳转化为有用的化学品和燃料成为一种可持续的能源解决方案。然而,目前存在的光催化材料在CO2还原反应中产生的产物主要是低值的一氧化碳或甲烷,而高值的C2+产物(如乙烯和乙醇)产率较低。因此,本研究旨在通过重掺杂高价态阳离子的金属氧化物形成固溶体的方法,改善光催化CO2还原反应的效率,并调控产物选择性,以产生更多的C2+产物。这一研究背景对于推动太阳能驱动的CO2还原反应的发展具有重要意义。
图文解析:
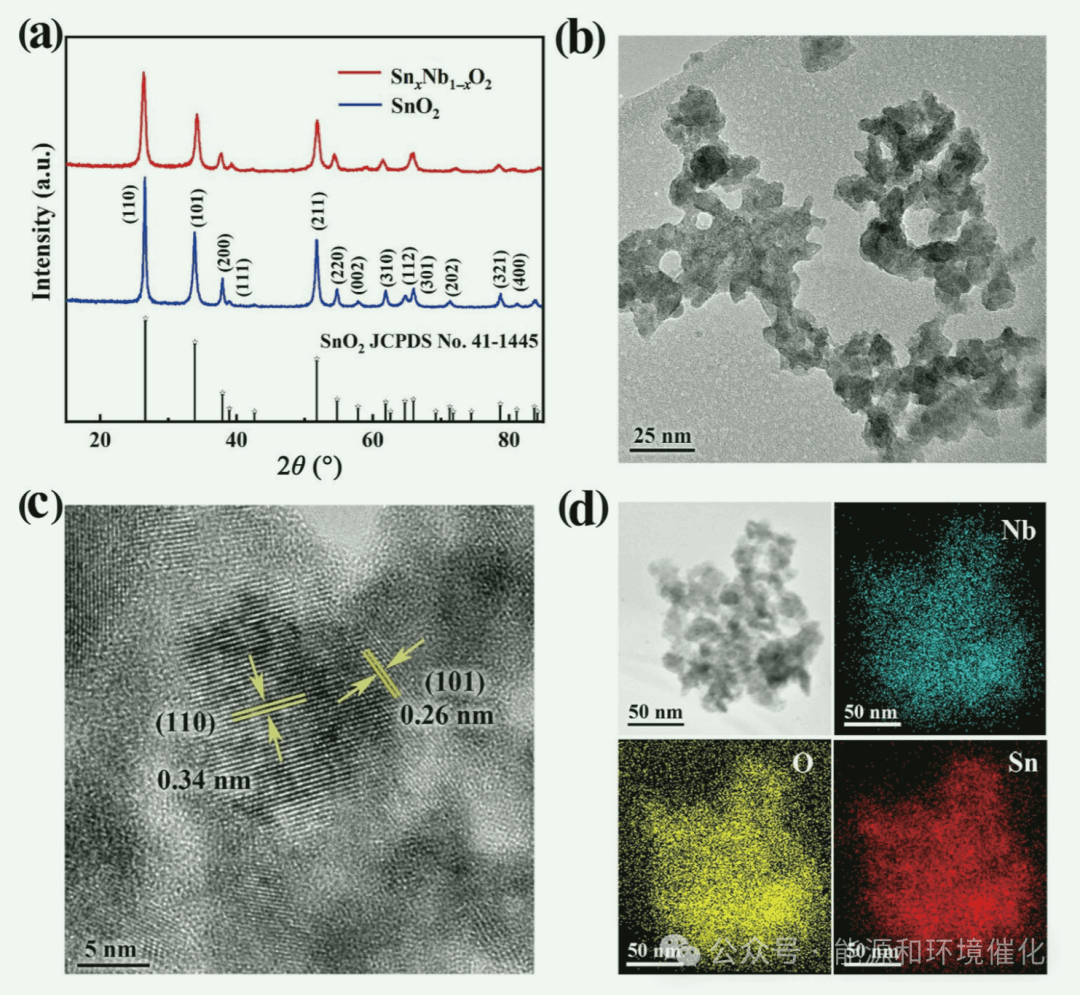
Fig. 1 (a) XRD patterns of SnxNb1−xO2 solid solution and pristine SnO2 samples. (b) Representative TEM image, (c) HRTEM image, and (d) element mappings of Nb, O, and Sn of the SnxNb1−xO2 solid solution sample.
图1(a)为SnxNb1−xO2固溶体和原始SnO2样品的XRD图谱。正如预期的那样,合成的SnO2样品具有金红石型晶体结构(JCPDS No. 41-1445)。对于SnxNb1−xO2固溶体,虽然其Nb: Sn原子比达到了~2.17,但其晶体结构仍与原始SnO2样品具有相同的金红石型晶体结构,并且没有观察到属于氧化铌的衍射峰。以SnO2为溶剂提供金红石结构晶格,Nb5+离子代替Sn4+形成所需的SnxNb1−xO2固溶体,合成了高Nb: Sn比的SnxNb1−xO2固溶体。图1(b)为SnxNb1−xO2固溶体样品的代表性TEM图像,可以看出SnxNb1−xO2固溶体样品由形状不规则的纳米颗粒组成,平均粒径为~15 nm。如此细的颗粒尺寸有利于获得具有活性位点的相对高的表面积。根据Bruauer - Emmet - Teller (BET)分析结果,测定其比表面积为~153.73 m2/g,而原始SnO2样品的比表面积为~50.98 m2/g。这些结果清楚地表明,通过重铌掺杂形成SnxNb1−xO2固溶体,大大提高了其比表面积,有利于其吸附和与CO2分子的反应。图1(c)为其高分辨率透射电子显微镜(HRTEM)图像。可以清楚地观察到两组晶格面,其d间距分别为~0.34和~0.26 nm,与金红石相的(110)和(101)晶格面匹配良好。这一观察结果与XRD分析结果一致,进一步验证了所合成的SnxNb1−xO2固溶体样品在水热反应下是高度结晶的。图1(d)为其EDS图谱结果,显示Nb、O、Sn元素在样品中均匀分布,这与我们的方法形成SnxNb1−xO2固溶体的事实相一致。
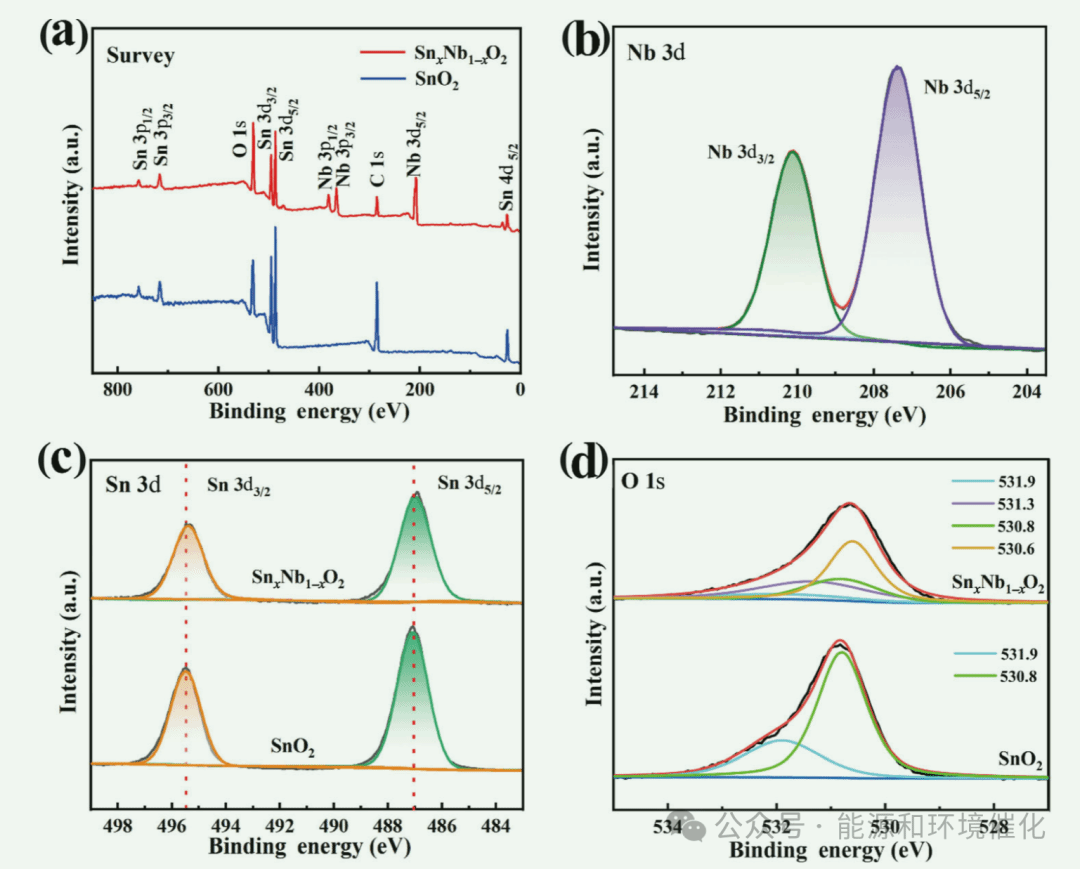
Fig. 2 (a) XPS survey spectra of SnxNb1−xO2 solid solution and pristine SnO2 samples. (b) High-resolution XPS scan over Nb 3d peaks of the SnxNb1−xO2 solid solution sample. (c, d) High-resolution XPS scans over Sn 3d and O 1s peaks of SnxNb1−xO2 solid solution and pristine SnO2 samples, respectively.
图2(a)为SnxNb1−xO2固溶体和原始SnO2样品的代表性XPS调查光谱。正如预期的那样,在两种样品中都可以发现Sn和O的信号,而Nb只能在SnxNb1−xO2固溶体样品中检测到。对于这两个样品,由于环境中碳的广泛存在,可以观察到C 1s峰。图2(b)为SnxNb1−xO2固溶体样品的Nb 3d峰的高分辨率XPS扫描图,其中Nb 3d3/2和Nb 3d5/2峰分别位于~210.1和~207.4 eV,符合Nb在五价氧化态的标准结合能。图2(c)比较了两个样品在Sn 3d峰上的高分辨率XPS扫描。SnxNb1−xO2固溶体样品的Sn4+ 3d3/2 (495.4 eV)和Sn4+ 3d5/2 (487.1 eV)峰与SnO2原始样品相比,均有轻微的低结合能方向偏移。这表明SnxNb1−xO2固溶体样品的电导率增加是由于Sn4+被Nb5+取代后载流子浓度增加,从而提高了与光催化反应相关的表面活性。图2(d)比较了两个样品在O 1s峰上的高分辨率XPS扫描。对于原始SnO2样品,其O 1s峰可以拟合为~531.9和~530.8 eV两个峰的组合,分别属于吸附的O物质和晶格Sn-O。对于SnxNb1−xO固溶体样品,其O 1s峰可以拟合为~531.9、~531.3、~530.8和~530.6 eV的4个峰的组合,分别代表吸附的O物质、氧空位、晶格Sn-O和晶格Nb-O。
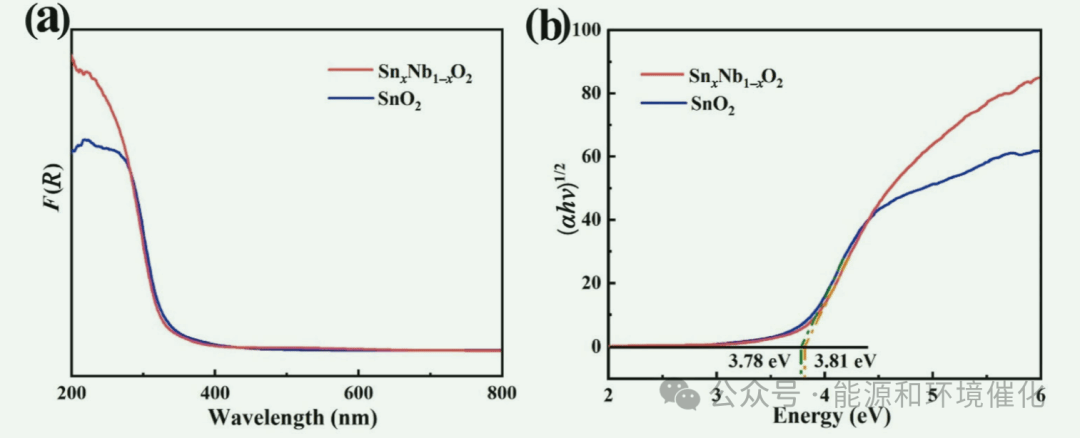
Fig. 3 (a) Light absorbance spectra of SnxNb1−xO2 solid solution and pristine SnO2 samples. (b) Their Tauc plot curves ((F(R)hv)0.5 vs. hv) constructed from their light absorbance data in Fig. 3(a).
图3(a)比较了SnxNb1−xO2 固溶体和原始SnO2样品的光吸收光谱,可以看出它们具有相似的光学性质。两者的主要吸光度都在紫外光范围内,但SnxNb1−xO2固溶体样品的吸光度强度高于原始SnO2样品。图3(b)显示了它们的Tauc图((F(R)hv)0.5 vs. hv),它们的带隙值可以通过将Tauc图的线性部分外推到光子能量的横坐标来确定。结果表明,SnxNb1−xO2固溶体和原始SnO2样品的带隙值分别为~ 3.81 eV和~ 3.78 eV,表明SnO2的重铌掺杂对其带隙值没有明显影响。
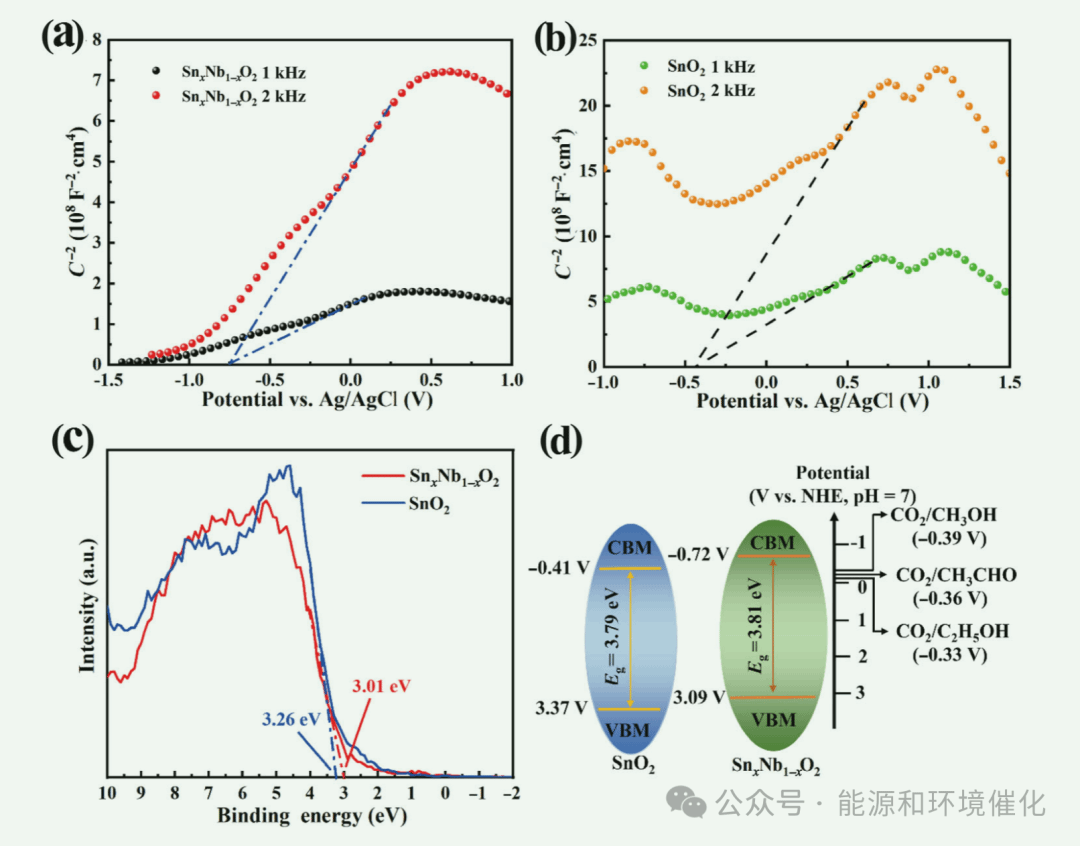
Fig. 4 M–S plots of (a) SnxNb1−xO2 solid solution and (b) pristine SnO2 samples vs. the Ag/AgCl reference electrode measured at 1 and 2 kHz, respectively. (c) XPS valence band spectra of SnxNb1−xO2 solid solution and pristine SnO2 samples. (d) Energy band structure diagrams of SnxNb1−xO2 solid solution and pristine SnO2 samples.
图4(a)和图4(b)分别显示了SnxNb1−xO2固溶体和原始SnO2样品与在1和2 kHz下测量的Ag/AgCl参比电极的M-S图。M-S曲线均呈现正斜率,说明SnxNb1−xO2固溶体的形成并未改变其n型半导体性质。从它们的M-S图的截距可以计算出它们相对于Ag/AgCl电极的平带电位,SnxNb1−xO2固溶体样品为~−0.74 V,原始SnO2样品为~−0.43 V。因此,它们相对于正常氢电极(NHE)的平带电位可以计算为:SnxNb1−xO2固溶体样品为~−0.52 V,原始SnO2样品为~−0.21 V。对于n型半导体,它们的平带电位等于它们的费米能级,通常比它们的导带最小值(CBMs)高~0.2 V。因此,SnxNb1−xO2固溶体样品的CBMs可以在~−0.72 V和原始SnO2样品的~−0.41 V下测定。因此,结合它们的CBMs和带隙值,可以计算出SnxNb1−xO2固溶体样品的价带最大值(VBMs)为~3.09 V,原始SnO2样品的价带最大值为~3.37 V。图4(c)为它们的XPS价带光谱。结果表明,SnxNb1−xO2固溶体样品的价带最大值为~3.01 eV, SnO2原始样品的价带最大值为~3.26 eV,这与带隙值和M-S图的结果一致。图4(d)是根据这些分析结果构建的它们的能带结构图。它们的CBM值均大于CO2/CH3OH (- 0.39 V)、CO2/CH3CHO (- 0.36 V)和CO2/C2H5OH (- 0.33 V)的还原电位,表明它们在光催化还原CO2为CH3OH、CH3CHO和C2H5OH方面是可行的。虽然两种样品具有相似的光学性质和带隙值,但SnxNb1−xO2固溶体的形成确实改变了SnO2的能带结构。特别是SnxNb1−xO2固溶体样品(~−0.72 V)的CBM值比原始SnO2样品(~−0.41 V)负0.31 V,这可以显著增强其光生电子的还原电位,从而提高其光催化CO2还原性能。
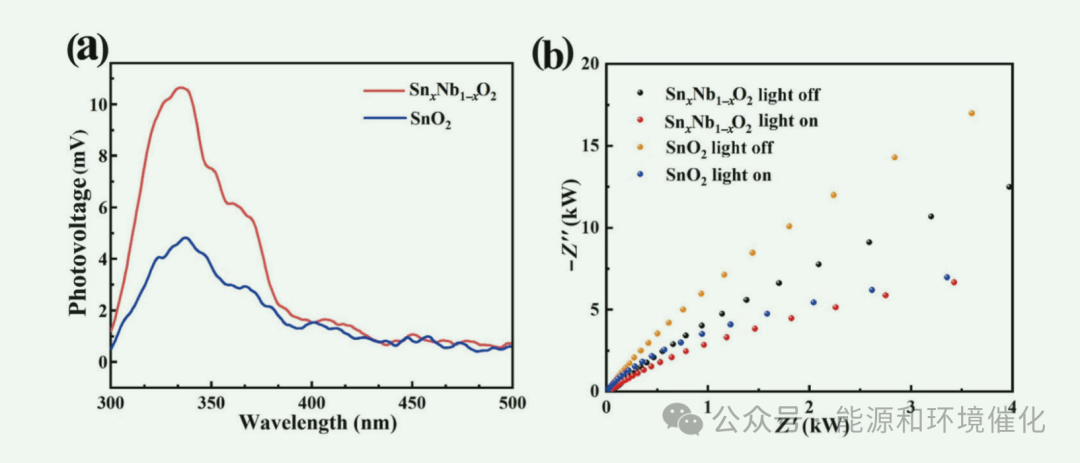
Fig. 5 (a) SPS of SnxNb1−xO2 solid solution and pristine SnO2 samples. (b) Nyquist plots of SnxNb1−xO2 solid solution and pristine SnO2 samples in darkness and under simulated solar illumination.
图5(a)显示了获得的样品的SPS,以研究在模拟太阳光照下其光生载流子的分离和转移。SnxNb1−xO2固溶体样品的表面光电压明显高于原始SnO2样品,这表明重铌掺杂形成SnxNb1−xO2固溶体由于氧空位的形成,可以显著促进光生载流子的分离和转移,从而提高其光催化性能。图5(b)比较了黑暗和模拟太阳光照下EIS测量得到的奈奎斯特图。奈奎斯特曲线的弧半径表示电极表面发生的反应速率;因此,较小的弧半径表示光生电荷载流子的分离水平较高,并且界面电荷更快地转移到电子受体或供体。在黑暗中,SnxNb1−xO2固溶体样品的圆弧半径比原始SnO2样品小,这证实了Nb5+取代Sn4+增加了其载流子浓度。在模拟太阳光照下,两种样品的电弧半径均因光照产生载流子而减小,而SnxNb1−xO2固溶体样品的电弧半径也比原始SnO2样品小。因此,通过重铌掺杂形成SnxNb1−xO2固溶体,确实提高了光生电荷的分离和转移速率,这与SPS测量结果一致,有利于提高其光催化性能。
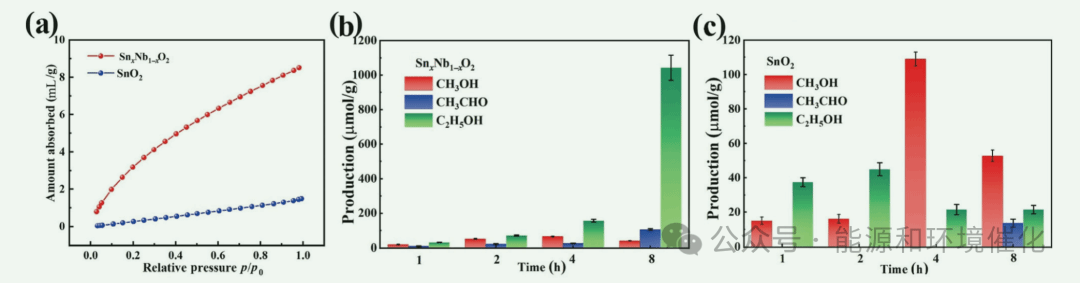
Fig. 6 (a) CO2 adsorption curves of SnxNb1−xO2 solid solution and pristine SnO2 samples. (b, c) Product evolution of photocatalytic CO2 reduction by SnxNb1−xO2 solid solution and pristine SnO2 samples under simulated solar illumination for a series of reaction time from 1 to 8 h, respectively.
图6(a)显示了SnxNb1−xO2固溶体和原始SnO2样品对CO2的吸附性能。在整个相对压力范围内,由于Sn4+被Nb5+取代,SnxNb1−xO2固溶体表面对CO2的吸附始终强于原始SnO2表面。例如,当相对压力为1.0时,SnxNb1−xO2固溶体样品的CO2吸附量达到~8.565 mL/g,是原始SnO2样品(~1.497 mL/g)的~5.7倍。为避免表面积效应,CO2吸附数据采用相应的表面积归一化处理。相对压力为0.0时,SnxNb1−xO2固溶体样品的CO2吸附量达到~0.056 mL/g,仍是原始SnO2样品(~0.029 mL/g)的~1.9倍。CO2分子在光催化剂表面的吸附是光催化CO2还原的第一步。因此,可以预期SnxNb1−xO2固溶体样品比原始SnO2样品具有更好的光催化CO2还原性能。与之前的报道不同,光催化CO2还原实验中没有使用牺牲剂,这可以大大降低操作成本/复杂性,有利于其潜在的实际应用。图6(b)为模拟太阳光照下SnxNb1−xO2固溶体样品在1 ~ 8 h的一系列反应时间下光催化CO2还原的产物演变。图6(c)为相同反应条件下原始SnO2样品的产物演化。两种样品在模拟太阳光照下的光催化CO2还原产物均为CH3OH、CH3CHO和C2H5OH的液体产物,未发现CO和CH4等气体产物。这一观察结果与以往报道的SnO2基光催化剂光催化CO2还原主要生成CO和CH4有很大不同。虽然SnxNb1−xO2固溶体样品与原始SnO2样品具有相似的光催化CO2还原产物,但它们的性能却有很大差异。SnxNb1−xO2固溶体样品比原始SnO2样品具有更好的光催化CO2还原效率。例如,模拟太阳光照8 h后SnxNb1−xO2固溶体样品的CH3OH、CH3CHO和C2H5OH的产率分别达到~42.0µmol/g、~106.0µmol/g和~1043.0µmol/g,而原始SnO2样品的CH3OH、CH3CHO和C2H5OH的产率分别为~52.8µmol/g、~13.7µmol/g和~21.5µmol/g。因此,经过8 h处理后,SnxNb1−xO2固溶体样品的CO2总还原量达到~2340.0µmol/g,是原始SnO2样品(~123.2µmol/g)的19倍。
结论:
综上所述,本文提出了一种新的缺陷工程方法,通过一步水热法制备SnO2重取代Nb掺杂SnxNb1−xO2固溶体。在不需要牺牲剂的情况下,大大提高了模拟太阳光照下光催化CO2还原性能。SnxNb1−xO2固溶体的形成提高了其导带和价带,显著提高了其光生电子的还原电位,降低了其光生空穴的氧化电位。此外,通过重铌掺杂,促进了光生载流子的分离和转移,增强了CO2分子在光催化剂表面的吸附,并在其表面形成了丰富的酸中心和多余电子的存在。所有这些因素都有利于光催化CO2还原,因此SnxNb1−xO2固溶体的光催化CO2还原效率是原始SnO2样品的19倍,并且对优选的C2+产物有良好的选择性。本研究表明,在金属氧化物中掺杂高价阳离子形成固溶体可能是一种很有前途的缺陷工程方法,可以增强光催化CO2还原和调节其还原过程以产生理想的C2+产物。这可以很容易地应用于各种光催化剂系统,以推进太阳能驱动的二氧化碳减排的发展。
